


中国罕见病综合报告(2021)
- 作者:陈懿玮 李杨阳
- 来源:知乎陈懿玮Echo
- 发布时间:2021-11-18 10:11

中国罕见病综合报告(2021)
【概要描述】从全球到中国,从医者到患者,从绝境中创新,从过去到未来……以数据勾勒现实,写在国际罕见病日之际。
- 作者:陈懿玮 李杨阳
- 来源:知乎陈懿玮Echo
- 发布时间:2021-11-18 10:11

全球视角
01 何为罕见病?
“罕见病”一词译自英语rare disease。在全球权威的生物医学数据库PubMed中,rare disease一词最早出现在1867年。

与“常见病”相对,“罕见病”指患病率相对较低的一类疾病。
欧盟地区定义“罕见病”的标准是患病率<1/2000,台湾地区的标准是<1/10000。另一些国家则根据全国患者的数量来界定,比如在日本,患者<5万人的疾病被认为是“罕见病”。
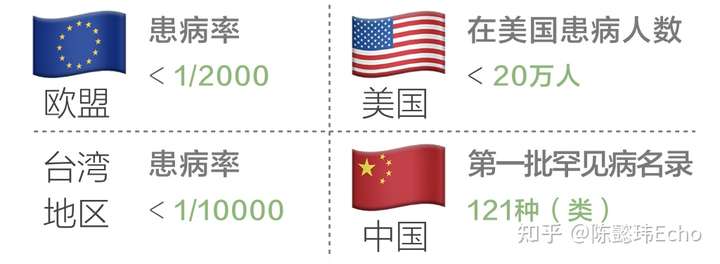
作为最早提出“罕见病”概念的国家,美国在1983年颁布的《孤儿药法案》中提出双重定义:
- 在美国患者人数<20万;
- 患者人数≥20万,但针对该疾病所开发的药物销售额不足以抵偿研发以及上市成本。
不同的国家/地区所划定“罕见病”的范围因国情而异,并不存在全球统一的患病率标准。“罕见病”的定义并非只为解决医学问题,更是为应对威胁人类健康的公共卫生问题提供政策制定的范围与依据。
02 罕见的困境
截止2018年10月,全球最大的罕见病数据库Orphanet共收录6172种罕见病,约占全部人类疾病的10%。其中,约72%的罕见病是遗传物质的结构改变或调控异常造成的,也就是遗传病。
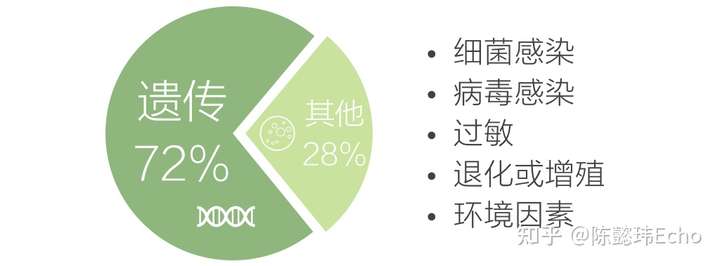
罕见病对人体的影响通常涉及多系统、多脏器;病程往往呈慢性、进行性、耗竭性地发展,甚至造成残疾或危及生命。在Orphanet数据库中,5018种(81.3%)罕见病记录了发病时间的数据,其中:
- 3510种仅在儿童期发病,占56.9%;
- 600种仅在成年期发病,占9.7%;
- 908种从儿童期到成年期皆可发病,占14.7%。
由于每种罕见病的患者数量少且信息匮乏,使得罕见病药物的开发困难重重——研发成本高昂、市场空间狭小、投资回报率低但风险高,因此乏人问津。罕见病药物有一个令人闻之心酸的别名——“孤儿药”。
为激励企业投入孤儿药研发,美国在1983年颁布《孤儿药法案》,出台一系列激励政策,包括对孤儿药给予优先审批、市场独占权、税收减免、研发支持等。
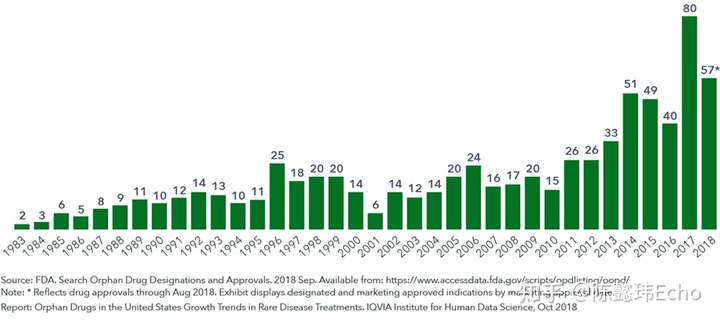
截止2018年8月,美国食品药品管理局(FDA)已批准上市503种孤儿药,针对731种罕见病适应症,约占所有罕见病的10%。
03 不罕见的罕见病人
全球每时每刻有多少患者正在直面罕见病的挑战呢?2.63 - 4.46 亿患者!全球罕见病患者的总数是惊人的,超过了全部癌症与艾滋病患者的总和。假如全球的罕见病患者组成一个国家,那将会是世界第三人口大国,仅次于中国与印度,与美国不相上下。
这同时意味着,我们身边每17-29个人中,就有 1 位正被某一种罕见病折磨着。不幸的是,这些患者中有一半是儿童。更不幸的是,这些孩子中有 30% 无法活着庆祝自己的 5 岁生日。在全球各地,罕见病的诊疗可能都是人类目前所面临的最大的医学挑战。
聚焦中国
04 国家定义篇
2018年5月,国家卫健委等五部委联合发布《第一批罕见病目录》,共收录121种(类)罕见病 :
- 21-羟化酶缺乏症 (21-Hydroxylase Deficiency)
- 白化病 (Albinism)
- 遗传性进行性肾炎 / Alport 综合征 (Alport Syndrome)
- 肌萎缩侧索硬化症 (Amyotrophic Lateral Sclerosis)
- Angelman 综合征 (Angelman Syndrome)
- 精氨酸酶缺乏症 (Arginase Deficiency)
- 窒息性胸腔失养症 / Jeune 综合征 (Asphyxiating Thoracic Dystrophy / Jeune Syndrome)
- 非典型溶血性尿毒症 (Atypical Hemolytic Uremic Syndrome)
- 自身免疫性脑炎 (Autoimmune Encephalitis)
- 自身免疫性垂体炎 (Autoimmune Hypophysitis)
- 自身免疫性胰岛素受体病 (Autoimmune Insulin Receptopathy)
- β-酮硫解酶缺乏症 (β-Ketothiolase Deficiency)
- 生物素酶缺乏症 (Biotinidase Deficiency)
- 心脏离子通道病 (Cardic Ion Channelopathies)
- 原发性肉碱缺乏症 (Carnitine Deficiency)
- Castleman 病 (Castleman disease)
- 腓骨肌萎缩症(Charcot-Marie-Tooth Disease)
- 瓜胺酸血症 (Citrullinemia)
- 先天性肾上腺发育不良 (Congenital Adrenal Hypoplasia)
- 先天性高胰岛素性低血糖血症 (Congenital Hperinsulinemic Hypoglycemia)
- 先天性肌无力综合征 (Congenital Myasthenia Syndrome)
- 先天性肌强直 (Congenital Myotonia Syndrome / Non-Dystrophic Myotonia)
- 先天性脊柱侧弯 (Congenital Scoliosis)
- 冠状动脉扩张病 (Coronary Artery Ectasia)
- 先天性纯红血球再生障碍性贫血 (Diamond-Blackfan Anemia)
- Erdheim-Chester 病 (Erdheim-Chester Disease)
- Fabry 病 (Fabry Disease)
- 家族性地中海热 (Familial Mediterranean Fever)
- 范可尼贫血 (Fanconi Anemia)
- 半乳糖血症 (Galactosemia)
- 戈谢病 (Gaucher Disease)
- 全身型重症肌无力 (General Myasthenic Gravis)
- Gitelman 综合征 (Gitelman Syndrome)
- 戊二酸血症 I 型 (Glutaric Acidemia Type 1)
- 糖原累积病(Ⅰ型、Ⅱ型) {Glycogen Storage Disease (Type 1、2)}
- 血友病 (Hemophilia)
- 肝豆状核变性/Wilson 病 (Hepatolenticular Degeneration / Wilson Disease)
- 遗传性血管性水肿 (Hereditary Angioedema)
- 遗传性大疱性表皮松解症 (Hereditary Epidermolysis Bullosa)
- 遗传性果糖不耐受症 (Hereditary Fructose Intolerance)
- 遗传性低镁血症 (Hereditary Hypomagnesemia)
- 遗传性多发脑梗死性痴呆 (Hereditary Multi-Infarct Dementia / Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts And Leukoencephalopathy)
- 遗传性痉挛性截瘫 (Hereditary Spastic Paraplegia)
- 全羧化酶合成酶缺乏症 (Holocarboxylase Synthetase Deficiency)
- 同型半胱氨酸血症 (Homocysteinemia)
- 纯合子家族性高胆固醇血症(Homozygous Hypercholestrolemia)
- 亨廷顿病 (Huntington Disease)
- HHH 综合征 (Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome)
- 高苯丙氨酸血症 (Hyperphenylalaninemia)
- 低碱性磷酸酶血症 (Hypophosphatasia)
- 低磷性佝偻病 (Hypophosphatemic Rickets)
- 特发性心肌病 (Idiopathic Cardiomyopathy)
- 特发性低促性腺激素性性腺功能减退症 (Idiopathic Hypogonadotropic Hypogonadism)
- 特发性肺动脉高压 (Idiopathic Pulmonary Arterial Hypertension)
- 特发性肺纤维化 (Idiopathic Pulmonary Fibrosis)
- IgG4 相关性疾病 (IgG4-related Disease)
- 先天性胆汁酸合成障碍 (Inborn Errors of Bile Acid Synthesis)
- 异戊酸血症(Isovaleric Acidemia)
- 卡尔曼综合征 (Kallmann Syndrome)
- 朗格汉斯细胞组织细胞增生症 (Langerhans Cell Histiocytosis)
- 莱伦氏综合征 (Laron Syndrome)
- Leber 遗传性视神经病变 (Leber Hereditary Optic Neuropathy)
- 长链3-羟酰基辅酶A脱氢酶缺乏症 (Long Chain 3-hydroxyacyl-CoA Dehydrogenase Deficiency)
- 淋巴管肌瘤病 (Lymphangioleiomyomatosis)
- 赖氨酸尿蛋白不耐受症 (Lysine Urinary Protein Intolerance)
- 溶酶体酸性脂肪酶缺乏症 (Lysosomal Acid Lipase Deficiency)
- 枫糖尿症 (Maple Syrup Urine Disease)
- Marfan 综合征 (Marfan syndrome)
- McCune-Albright 综合征 (McCune-Albright Syndrome)
- 中链酰基辅酶A脱氢酶缺乏症 (Medium Chain Acyl-Coa Dehydrogenase Deficiency)
- 甲基丙二酸血症 (Methylmalonic Acidemia)
- 线粒体脑肌病 (Mitochondrial Encephalopathy)
- 黏多糖贮积症 (Mucopolysaccharidosis)
- 多灶性运动神经病 (Multifocal Motor Neuropathy)
- 多种酰基辅酶A脱氢酶缺乏症 (Multiple Acyl-CoA Dehydrogenase Deficiency)
- 多发性硬化症 (Multiple Sclerosis)
- 多系统萎缩 (Multiple System Atrophy)
- 肌强直性营养不良 (Myotonic Dystrophy)
- N-乙酰谷氨酸合成酶缺乏症 (NAGS Deficiency)
- 新生儿糖尿病 (Neonatal Diabetes Mellitus)
- 视神经脊髓炎 (Neuromyelitis Optica)
- 尼曼匹克病 (Niemann-Pick Disease)
- 非综合征性耳聋 (Non-Syndromic Deafness)
- Noonan 综合征 (Noonan Syndrome)
- 鸟氨酸氨甲酰基转移酶缺乏症 (Ornithine Transcarbamylase Deficiency)
- 成骨不全 (Osteogenesis Imperfecta)
- 帕金森病(青年型、早发型){Parkinson Disease (Young-onset, Early-onset)}
- 阵发性睡眠性血红蛋白尿 (Paroxysmal Nocturnal Hemoglobinuria)
- 黑斑息肉综合征 (Peutz-Jeghers Syndrome)
- 苯丙酮尿症 (Phenylketonuria)
- POEMS 综合症 (POEMS Syndrome)
- 卟啉病 (Porphyria)
- Prader-Willi 综合征 (Prader-Willi Syndrome)
- 原发性联合免疫缺陷 (Primary Combined Immune Deficiency)
- 原发性遗传性肌张力不全 (Primary Hereditary Dystonia)
- 原发性轻链型淀粉样变 (Primary Light Chain Amyloidosis)
- 进行性家族性肝内胆汁淤积症 (Progressive Family Intrahepatic Cholestasis)
- 进行性肌营养不良 (Progressive Muscular Dystrophies)
- 丙酸血症 (Propionic Acidemia)
- 肺泡蛋白沉积症 (Pulmonary Alveolar Proteinosis)
- 肺囊性纤维化 (Pulmonary Cystic Fibrosis)
- 视网膜色素变性症 (Retinitis Pigmentosa)
- 视网膜母细胞瘤 (Retinoblastoma)
- 重症先天性粒细胞缺乏症 (Severe Congenital Neutropenia)
- 婴儿严重肌阵挛性癫痫 / Dravet 综合征 (Severe Myoclonic Epilepsy in Infancy / Dravet Syndrome)
- 镰刀型细胞贫血症 (Sickle Cell Disease)
- Silver-Russell 综合征 (Silver-Russell Syndrome)
- 谷固醇血症 (Sitosterolemia)
- 脊髓延髓肌萎缩症 / 肯尼迪病 (Spinal and Bulbar Muscular Atrophy / Kennedy Disease)
- 脊髓性肌萎缩症 (Spinal Muscular Atrophy)
- 脊髓小脑性共济失调 (Spinocerebellar Ataxia)
- 系统性硬化症 (Systemic Sclerosis)
- 四氢生物蝶呤缺乏症 (Tetrahydrobiopterin Deficiency)
- 结节性硬化 (Tuberous Sclerosis Complex)
- 原发性酪氨酸血症 (Tyrosinemia)
- 极长链酰基辅酶A脱氢酶缺乏症 (Very Long Chain Acyl-CoA Dehydrogenase Deficiency)
- 威廉姆斯综合征 (Williams Syndrome)
- 湿疹血小板减少伴免疫缺陷综合征 (Wiskott-Aldrich Syndrome)
- X-连锁无丙种球蛋白血症 (X-linked Agammaglobulinemia)
- X-连锁肾上腺脑白质营养不良 (X-linked Adrenoleukodystrophy)
- X-连锁淋巴增生症X-linked (Lymphoproliferative Disease)
同月,国家卫健委发布《罕见病目录制订工作程序》,将分批遴选目录覆盖病种,对目录进行动态更新;更新时间原则上不短于2年。纳入目录的病种应同时满足以下条件:
- 国内外有证据表明发病率或患病率较低;
- 对患者和家庭危害较大;
- 有明确诊断方法;
- 有治疗或干预手段、经济可负担,或尚无有效治疗或干预手段、但已纳入国家科研专项。
2019年2月,国家卫健委发布关于建立全国罕见病诊疗协作网的通知,首批遴选324家医院组建罕见病诊疗协作网,包括1家国家级、32家省级牵头医院和291家协作网成员医院。
同月,国家卫健委发布《罕见病诊疗指南(2019年版)》。10月,发布关于开展罕见病病例诊疗信息登记工作的通知,组织开发中国罕见病诊疗服务信息系统。
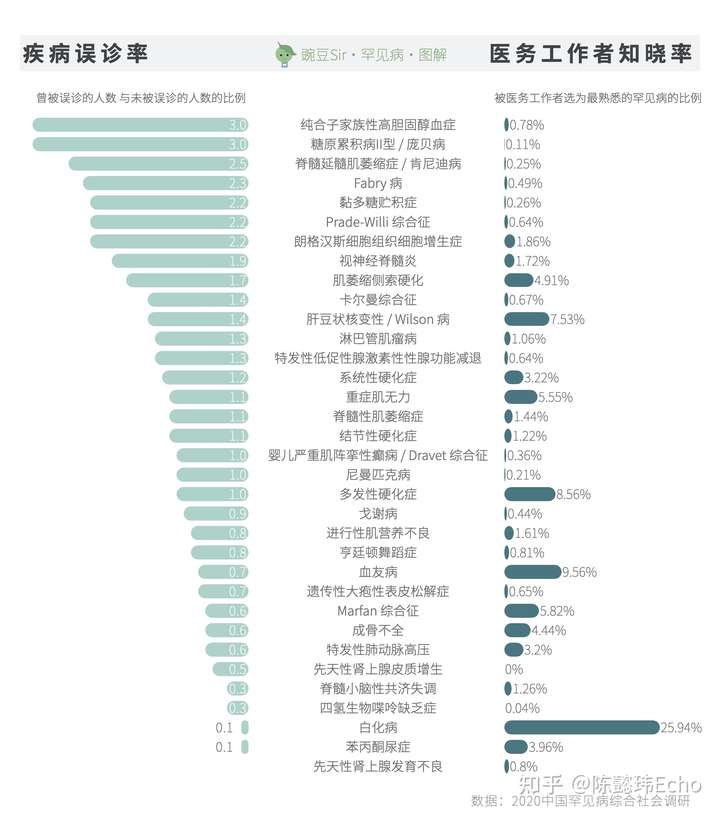
与之相对的是,针对38634名中国医务工作者的调查显示:
- 1770人从未听说过罕见病,占4.6%
- 23514人听说过但不了解,占60.9%。
其中,医务工作者最熟悉的罕见病依次是白化病、血友病和多发性硬化症。同时,87.6%的医务工作者认为自己并不了解国家关于罕见病的政策。
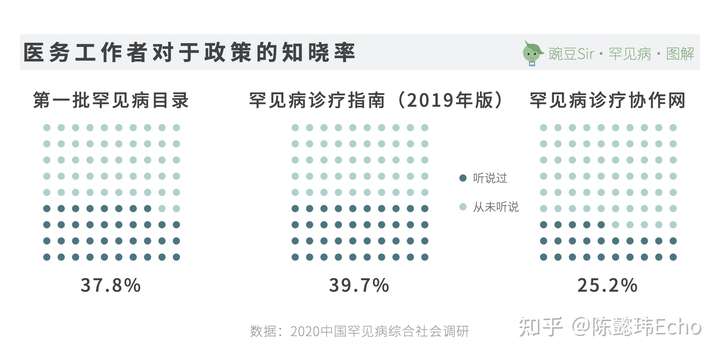
对罕见病较为了解、有诊治经验的医务工作者通常集中在大城市。由于优质医疗资源的分布不均,长途跋涉辗转异地就医是大多数中国患者的常态。
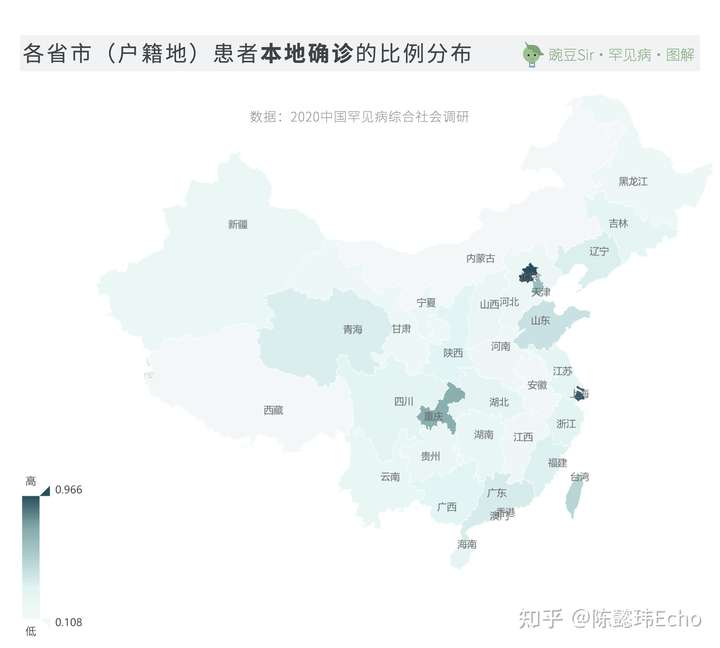
96.6%的北京患者、93.8%的上海患者和71.3%的重庆患者可以实现在本地确诊;而100%的西藏患者和83.7%的内蒙古患者则需要在省外异地确诊。
为改善罕见病的诊疗现状,自2020年3月起,在中国罕见病联盟的支持下,豌豆Sir与中国国家罕见病注册系统(NRDRS)专家合作,联合出品,旨在为广大一线的医务工作者提供全面、可靠、深入的罕见病知识,以及可实操的诊疗经验。
05 药物可及篇
随着药政改革的推进,国家药品监督管理局连续出台了三批《临床急需境外新药名单》,连同优先审评审批政策,对罕见病药品在我国上市起到极大的助推作用。
自2019年至今,以《第一批目录》作为定义依据,我国已新增批准上市14种罕见病药物,涉及9种罕见病适应症,带来了前所未有的罕见病新药上市浪潮:

但截至2021年2月26日,以《第一批目录》为统计依据,仍有16种罕见病的患者在我国面临“境外有药、境内无药”的困境:
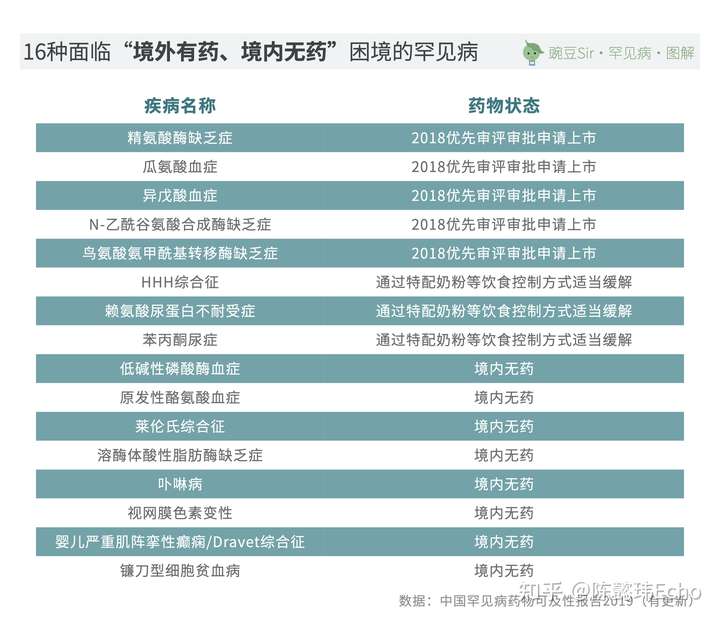
在《第一批目录》中,58种罕见病目前在我国有治疗药物上市,但其中有21种罕见病在使用的所有治疗药品皆为“超说明书”用药,这意味着无论是在医生处方还是患者自行购药时,都面临较大的不确定性,无法保障及时、可持续的治疗。

因此,严格意义上说,在我国,以《第一批目录》为统计依据,仅36种罕见病有“说明书内”用药,涉及68种治疗药物。
自2017年起,国家医保目录更新和谈判机制逐步常态化,每年有越来越多的罕见病药品被纳入医保。截至发稿,在我国已上市的68种罕见病药物中,有46种已被纳入国家医保目录,涉及23种罕见病适应症。
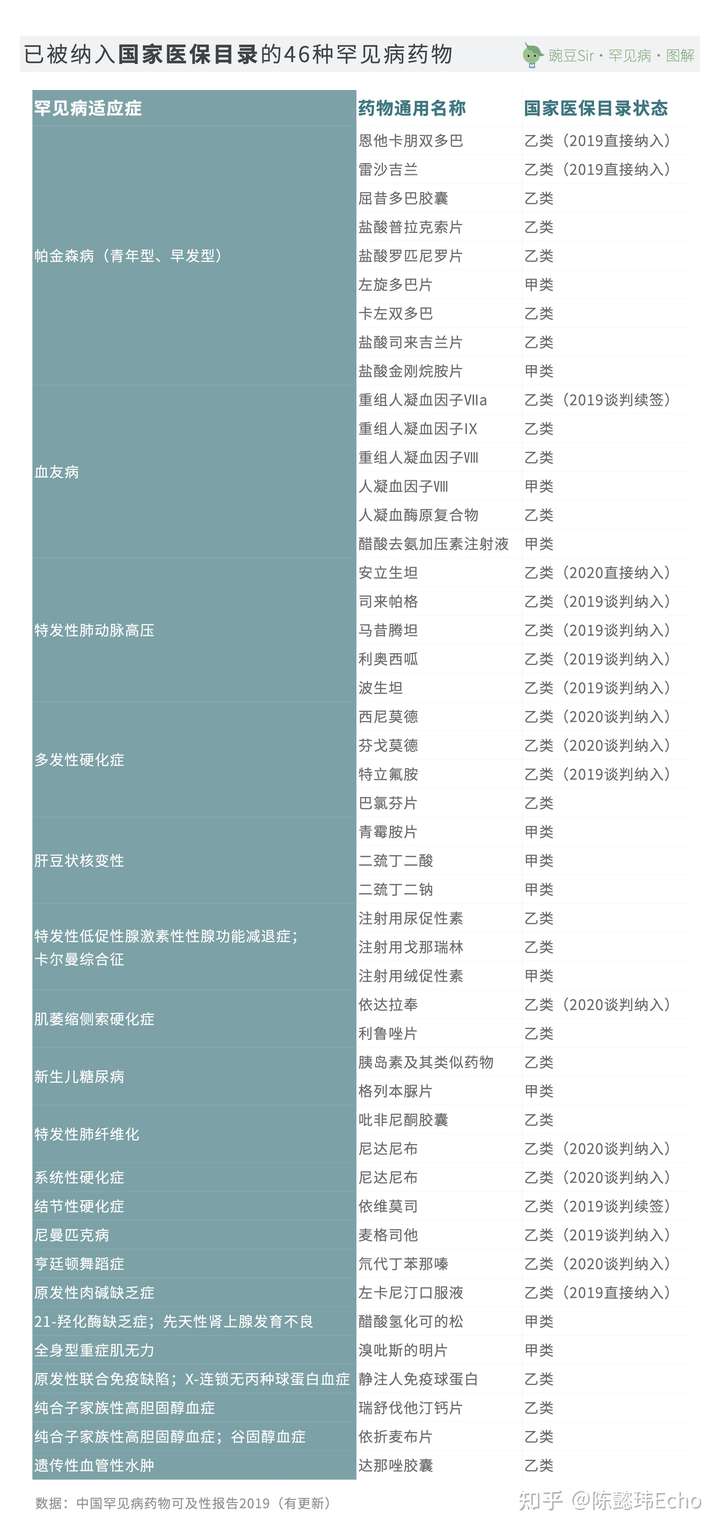
整体而言,我国对罕见病的可及已经实现了较大的进步,越来越多的罕见病药品被引进中国,大部分罕见病用药也已能够获得较好的保障。

目前,有13种罕见病在我国上市的所有治疗药物均未被纳入国家医保目录,涉及22种治疗药物;其中8种药物是对应5种罕见病的特效药物,年治疗费用皆超过百万元且需要患者终身维持使用——这正是目前社会关注的焦点,也是罕见病用药保障问题中矛盾最为突出的部分。
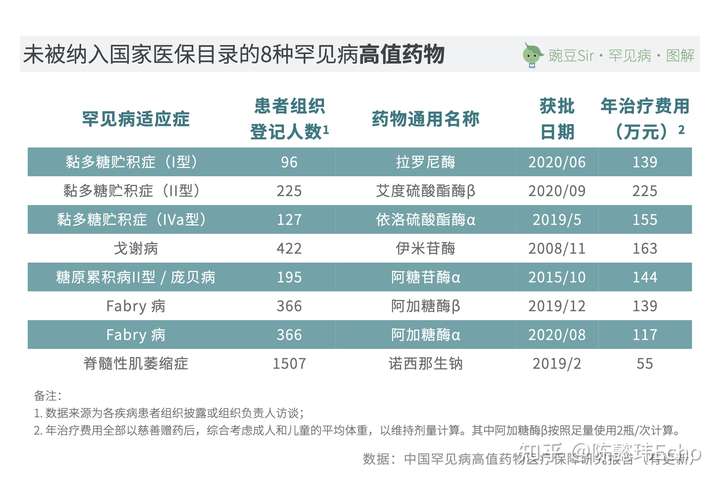
解决罕见病高值药物的用药保障,有着重要的社会意义和时代意义。《中国罕见病高值药物医疗保障研究报告》认为,中国有三种可探索的路径:
(1)“基本医疗保险+多层次保障”模式:在现有基本医疗保险的框架下,实现对罕见病高值药物保障的决策突破,让基本医疗保险率先发挥作用,带动多层次保障,实现对罕见病患者的用药保障。

(2)“国家制定目录准入标准+地方探索落地保障”模式:作为过渡方案,由国家医保局组织成立专家委员会并制定“罕见病高值药物保障清单”,由各省积极探索用药保障机制,为全国罕见病用药保障机制的建立积累经验、打好基础。
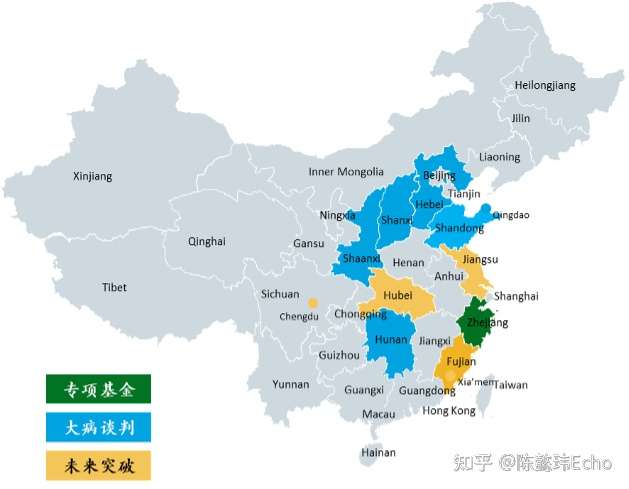
(3)“国家罕见病专项基金”模式:由国家统筹、专款专项、统一待遇水平、社会共付来实现对罕见病高值药的保障。
06 患者社群篇
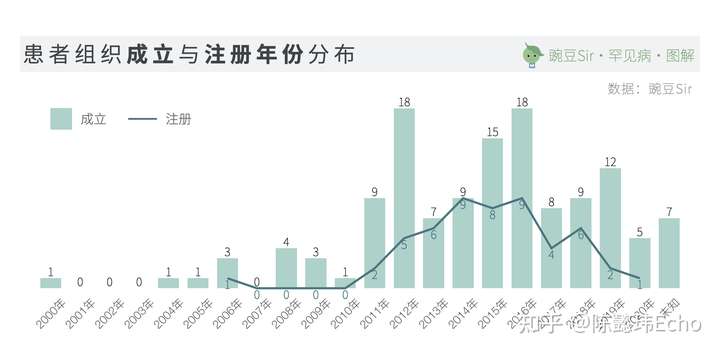
在全球,患者社群一直是推动罕见病政策制定、科研、制药、社会倡导的中坚力量。中国第一家罕见病患者组织“中国血友病联谊会”成立于2000年。截止目前,国内存在的名称固定、活动规律的罕见病患者组织已有130家:
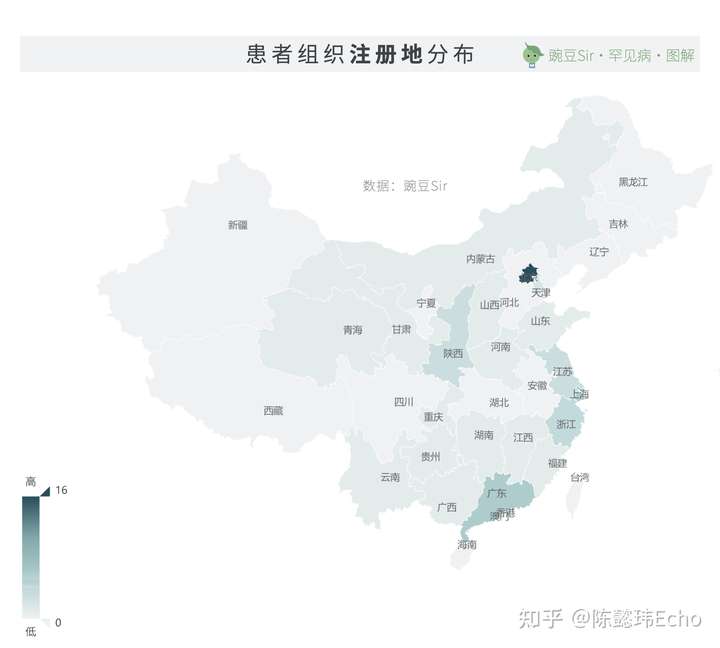
52家(40%)患者组织已在各地民政部门注册。其中,16家在北京市注册,数量最多;上海市与广东省各有6家。
89家(68.5%)患者组织所服务的所有疾病都已被纳入《第一批罕见病目录》;30家(23.1%)患者组织所服务的疾病未被纳入;11家(8.5%)组织兼而有之。37家(28.5%)患者组织仅由患者发起人;其余组织由家属共同发起。其中,17家(13.1%)由医生共同发起,5家(3.8%)由科研人员共同发起。
专业人士自2009年开始参与、共同组建患者组织,是非常令人欣喜的信号。不但标志着专业人士对于罕见病群体日益关注,更是引导患者群体走向专业化、职业化的重要推动。
在130家患者组织中,蚕宝儿LNS罕见病关爱之家是一个不同寻常的个例——起源于2018年3月“豌豆Sir”创作的Lesch-Nyhan 综合征的科普。近3年来,20多个家庭从全国各地循着科普而来,在“豌豆Sir”的帮助下:
- 2018年12月,成立患者组织;
- 2019年4月,网罗全国诊治过LNS的临床专家,组建中国LNS诊疗联盟;
- 2020年8月,召开首届诊疗联盟学术会议;
- 2020年12月,与国外LNS权威专家建立交流;
- 2021年2月,启动LNS卓越医学中心的全球合作。
然而,现实资源的制约与患者社群的活跃形成鲜明对比。在中国罕见病联盟针对74家罕见病组织的的调查中,63家没有固定的资金来源,占85.1%。
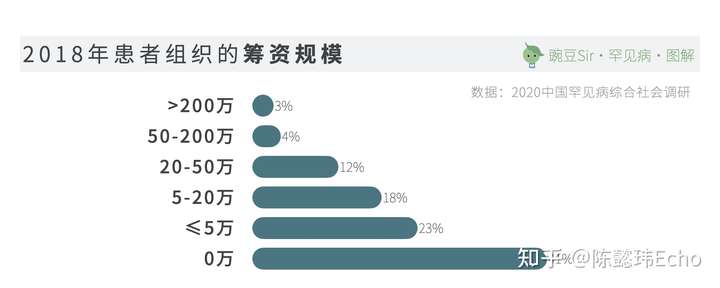
目前,患者组织的资金来源主要依赖组织内部成员的捐款,和企业捐赠/项目合作。作为罕见病生态中,至关重要的角色,罕见病社群亟需社会各界给予更多的支持与帮助。
参考资料:
- Watkins WH. Case of Rare Disease of Bone [J]. Glasgow Medical Journal, 1867, 2(18):233-235.
- Wakap SN, Lambert DM, Olry A, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database [J]. European Journal of Human Genetics, 2020, 28:165-173.
- IQVIA Institute. Orphan Drugs in the United States: Exclusivity, Pricing and Treated Populations [R] 2018.
- 国家卫生健康委员会. 关于公布第一批罕见病目录的通知:国卫医发〔2018〕10号 [A/OL]. 2018年5月11日.
- 国家卫生健康委员会. 关于印发罕见病目录制订工作程序的通知:国卫办医发〔2018〕11号 [A/OL]. 2018年5月28日.
- 国家卫生健康委员会. 关于建立全国罕见病诊疗协作网的通知:国卫办医函〔2019〕157号 [A/OL]. 2019年2月12日.
- 国家卫生健康委员会. 关于印发罕见病诊疗指南(2019年版)的通知:国卫办医函〔2019〕198号 [A/OL]. 2019年2月27日.
- 国家卫生健康委员会. 关于开展罕见病病例诊疗信息登记工作的通知:国卫办医函〔2019〕775号 [A/OL]. 2019年10月10日.
- 丁洁, 王琳. 中国罕见病研究报告(2018)[R]. 北京:中国医药科技出版社, 2018.
- 黄如方, 邵文斌. 中国罕见病药物可及性报告2019 [R]. 2019.
- 黄如方, 邵文斌. 中国罕见病医疗保障城市报告2020 [R]. 2020.
- IQVIA管理咨询团队. 中国罕见病高值药物医疗保障研究报告 [R]. 2020.
- 张抒扬, 董咚. 2020中国罕见病综合社会调研 [R]. 北京:人民卫生出版社. 2020.
责任编辑:亦欣
免责声明:本网注重分享,并不意味着赞同本文观点或证实内容的真实性,请仅做参考。著作权归原作者所有,在此向原作者表示感谢。除非无法确认,本网都会注明作者及来源。如有版权异义请及时告知。





")










